Previous work
-
Saturday 1st November 2014
-
Start from http://grade.globalphasing.org/tut/erice_workshop/1pmq_tutorial/
Question is the ligand charged?
-
Question arises because I think that it is best to use explicit hydrogen atoms on the ligand. So getting charge state correct is a good idea.
-
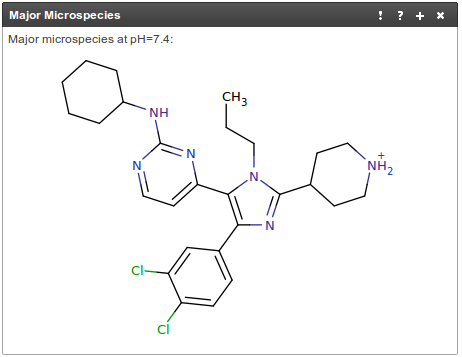
880 ligand image from RCSB:

-
From my (rather limited) chemistry I would expected the piperidine ring in 880 to be protonated at neutral pH. Scapin et al. (2003) give crystallization conditions pH 7.0 so charge it.
-
Lets check that this is correct (want a 2nd opinion).
-
Chemaxon http://www.chemicalize.org/ is exactly what is needed. Start with the PDB chemical components SMILE
CCCn1c(nc(c2ccc(Cl)c(Cl)c2)c1c3ccnc(NC4CCCCC4)n3)C5CCNCC5 -
Chemicalize says the ring is protonated:
-
In 1pmq the piperidine ring contacts phosphate of ANP modelled next to it - so the cationic piperidine makes sense.
-
-
So lets make a grade dictionary for the cationic form
-
The piperidine nitrogen atom is N19 and its existing hydrogen is H19 Lets call the extra hydrogen atom H192 position a pointer in coot and use to create 880_cation_initial.pdb. Conver to mol2 for grade use:
obabel 880_cation_initial.pdb -O880_cation_initial.mol2 grade -in 880_cation_initial.mol2 -ocif 880_cation.grade.cif -opdb 880_cation.grade.pdb -charge 1 -resname 880
-
Use resulting dictionary 880_cation.grade.cif for further work.
-
Model the alternate occupancy for the ligand
-
The dichlorophenyl ring clearly has two alternates. My prefence for modelling alternates is to model as few atoms as possible not the whole ligand (just for a protein side chain).
-
start from 1pmq_03_short.report/ model
-
add the extra hydrogen atom for the cation using hydrogenate
-
then look at http://www.globalphasing.com/buster/wiki//index.cgi?AutoBusterLigandAndOccRef
-
In coot markup alterate occupancy for atoms C35 C37 H37 C38 CL45 C39 CL46 C40 H40 C41 H41 using Measures, residue info, setting the occupancy for the heavy atoms to 0.6
-
tried in coot do a general torsion along C4 C35 to get an initial model of the B conformer but this did not work. So instead in an editor extract the alternate A atoms and modify to B change occupancy of heavy atoms to 0.4. 1pmq_03_cation880-altB.pdb. In coot rotate the ring 180 degrees around C4 C35 save coordinates.
-
In editor merge the B alternate coordinates. Results 1pmq_03_cation880-twoalts_init.pdb
-
-
For refinement will want to do occupancy refinement use pdb2occ to prepare a Gelly file
pdb2occ -p 1pmq_03_cation880-twoalts_init.pdb -o 03_occupancy_refine.Gelly
-
03_occupancy_refine.Gelly needed some manual adjustment as we have zero occupancy hydrogen atoms
-
-
refinement
-
script: 1pmq_04_ab_cation_two_alts.sh
-
results:
-
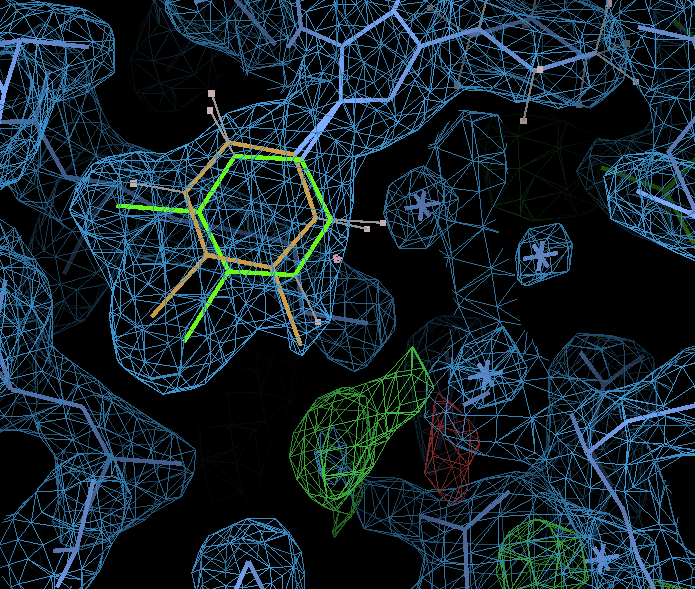
ED around the dichlorophenyl ring cleaned up by the alternates:
-
alternate A is shown in green and has occupancy 0.65 where is alternate B is brown with occupancy 0.35. Should place a water molecule in difference density below the ring. Picture also shows GLU 111 at the bottom that has clear difference density for two alternates (see below).
-
Occupancy second "accidental" AMP-PCP ligand Scapin et al. (2003) increases from 0.5 to 0.67. This fixes positive difference density around the triphosphate-N group that has really good density. Putting 2Fo-Fc contour level down to 0.8 , reasonable given the partial occupancy, shows weak but reasonable ED for the rest. Should try to re-fit the sugar. I am now convinced that the ANP placement is really correct - originally I had doubts! Lets leave any remodelling till the end in case ED is made clearer by other changes (see below).
-
-
-
Make table of re-refine progress.
run id |
01 |
02 |
03/04 |
description |
MapOnly run to analyse 1pmq.pdb (from tutorial) |
BUSTER refinement of 1pmq.pdb (from tutorial) |
remodel 880 ligand fixing cyclohexyl (03), then as cation with alternates, occupancy refinemnt |
script |
|||
buster-report |
|||
Rwork |
0.2231 |
0.2062 |
0.2044 |
Rfree |
0.2619 |
0.2362 |
0.2337 |
100*(Rfree-Rwork) |
3.9% |
3.0% |
2.9% |
Diffraction Anisotropy Server
-
Before doing anything further lets see where the anistropy server can help (as suggested by the tutorial).
-
start from 1pmq/1pmq.mtz
-
results dataAnsisotropyServer.pdf and dataAnsisotropyServer.mtz
-
05 run re-refine 04 using new mtzi. This results in Rwork/Rfree change from 0.2044/0.2337 to 0.2012/0.2305 (eliminates the worst reflections).
-
visually the map is slightly cleaner. Contouring new map at 1.5 rmsd gets contour matching volume 1.3 rmsd for 03. The density for side chains is better for instance for 49 TYR ED matches side chain. There is an unmodelled water H bonding to main chain 190 carbonyl. This appears in 2Fo-Fc at 2.0 rmsd compared to 1.1 rmsd previously.
-
subtle effect but worth having it.
run id |
01 |
02 |
03/04 |
05 |
description |
MapOnly run to analyse 1pmq.pdb (from tutorial) |
BUSTER refinement of 1pmq.pdb (from tutorial) |
remodel 880 ligand fixing cyclohexyl (03), then as cation with alternates, occupancy refinemnt |
use Diffraction Anisotropy Server |
script |
||||
buster-report |
||||
Rwork |
0.2231 |
0.2062 |
0.2044 |
0.2012 |
Rfree |
0.2619 |
0.2362 |
0.2337 |
0.2305 |
100*(Rfree-Rwork) |
3.9% |
3.0% |
2.9% |
2.9% |
MolProbity Score/percentile |
2.23 / 71st |
1.22 /100 |
1.30 / 100 |
1.43 / 100 |
MolProbity Poor Rotamer |
11 / 5.56% |
8 / 2.59% |
8 / 2.59% |
9 / 2.91% |
MolProbity Ramachandran outliers |
3 / 0.88% |
2 / 0.58% |
2 / 0.58% |
2 / 0.58% |
MolProbity Ramachandran favoured |
321 / 93.86% |
328 / 95.91% |
329 / 96.20% |
327 / 95.61% |
-
MolProbity scores get slightly worse with more refinement
General rebuild #1
-
Start from 05 structure/mtz
-
First lets check/build things close to the ligand.
-
GLU 111 A appears to be in two alternates. Tried modelling using coot split but do by modelling just and other and editing into a single pdb. Water 639 made alternate A and half occupancy as i it is where the B alternate is 1pmq_05_dataAnsisotropyServer-edit111.pdb.
-
Add a water molecule (HOH B 1) full occupancy H bonding between OE1 ASP 111 A conformer and NZ LYZ 93
-
Go through ED peaks (top already dealt with by B 1)
-
-
Place water B 2 between O 190, OD1 ASP 189 and HOH A 603.
-
Place water B 3 between N 125, HOH A 608 and HOH 629
-
Place water B 4 between N 96 and O 75
-
Place water B 6 between OD1 ASN 325 and N 163 and HOH A 605
-
Try to sort out N terminus moving ASN 46 and ASP 45. Got residues into ED but might be the wrong way around?
-
Call it a day and re-refine to check.
-
Alter new waters to chain A residues A 1001 to A 1006 to include in TLS
-
coordinates for re-refine 1pmq_05_dataAnsisotropyServer-edit111-coot-0-edit.pdb
-
-
re-refinement 1pmq_06_rebuild1.sh
-
results
-
the six new waters fine
-
ASP 111 alternates look good
-
N terminus 45 and 46 better than before. ED at 1 sigma. Blob could be waters or disorder protein. pusing it to interpret
-
progress table - keep 01, 02 and the last two .BUSTER re-refinements (up to 06)
-
run id |
01 |
02 |
05 |
06 |
description |
MapOnly run to analyse 1pmq.pdb (from tutorial) |
BUSTER refinement of 1pmq.pdb (from tutorial) |
use Diffraction Anisotropy Server |
place 6 waters, GLU 111 alts, N terminus |
script |
||||
buster-report |
||||
Rwork |
0.2231 |
0.2062 |
0.2012 |
0.2003 |
Rfree |
0.2619 |
0.2362 |
0.2305 |
0.2270 |
100*(Rfree-Rwork) |
3.9% |
3.0% |
2.9% |
2.7% |
MolProbity Score/percentile |
2.23 / 71st |
1.22 /100 |
1.43 / 100 |
1.43 / 100 |
MolProbity Poor Rotamer |
11 / 5.56% |
8 / 2.59% |
9 / 2.91% |
9 / 2.91% |
MolProbity Ramachandran outliers |
3 / 0.88% |
2 / 0.58% |
2 / 0.58% |
2 / 0.58% |
MolProbity Ramachandran favoured |
321 / 93.86% |
328 / 95.91% |
327 / 95.61% |
327 / 95.61% |
General rebuild #2
-
go through coot Density fit graph, remove side chain/residues with no evidence in ED (my prefered take):
-
52 GLU alter to mm-40 13% and real space fit.
-
55 ASP stub to CB
-
117 CYS and 118 LYS stub to CB
-
211 ALA and 212 ARG residues before an existing break. No ED at 1.0 rmsd remove both residues.
-
217 SER + 218 PHE weak ED at 0.8 sigma leave alone
-
219 MET sidechain no ED - stub to CB
-
220 MET weak ED at 0.8 sigma leave alone
-
221 THR + 222 PRO + 223 THR + 224 VAL no ED at 0.8 sigma remove all four residues.
-
225 VAL stub to CB
-
266 ARG stub to CB
-
288 LYS stub to CB
-
291 GLN stub to CB
-
295 ARG stub to CB
-
300 GLN stub to CB
-
301 ARG stub to CB
-
303 LYS stub to CB
-
319 PRO no density at 1.0 rmsd remove residue completely, extend to 320 ALA
-
321 ASP stub to CB
-
372 PRO and 373 PRO weak density at 0.8 rms leave alone
-
374 GLN remove residue no ED at 0.8
-
379 to 383 ED is weak remove these 5 residues
-
396 LYS stub to CB
-
400 ASN stub to CB
-
-
result of all this chopping: 1pmq_06_rebuild1-coot-3.pdb
-
re-refine this:
-
*On first attempt turned out had remove side chains of 163 HIS and 257 VAL by mistake (coot delete in blackspace!).
-
redo the run having restored these two side chains 1pmq_06_rebuild1-coot-4.pdb
-
Number of ATOM/HETATMs in this file is 2815 compared to 2977 before (removed ~5% of the structure).
-
-
re-refine as 07
run id |
01 |
02 |
06 |
07 |
description |
MapOnly run to analyse 1pmq.pdb (from tutorial) |
BUSTER refinement of 1pmq.pdb (from tutorial) |
place 6 waters, GLU 111 alts, N terminus |
chop out side chains and complete residues with no ED |
script |
||||
buster-report |
||||
Rwork |
0.2231 |
0.2062 |
0.2003 |
0.2027 |
Rfree |
0.2619 |
0.2362 |
0.2270 |
0.2298 |
100*(Rfree-Rwork) |
3.9% |
3.0% |
2.7% |
2.7% |
MolProbity Score/percentile |
2.23 / 71st |
1.22 /100 |
1.43 / 100 |
1.13 / 100 |
MolProbity Poor Rotamer |
11 / 5.56% |
8 / 2.59% |
9 / 2.91% |
6 / 2.13% |
MolProbity Ramachandran outliers |
3 / 0.88% |
2 / 0.58% |
2 / 0.58% |
0 / 0% |
MolProbity Ramachandran favoured |
321 / 93.86% |
328 / 95.91% |
327 / 95.61% |
312 / 96.59% |
-
So Rfree is up by 0.3% this is the same amount as placing 6 clear water molecules. Given that ~150 atoms that have weak density have been removed I think the cleaner structure justifies the change. Given more time I would look for the highest resolution/best JNK3 structure in this cell and try target restraints but this is not possible.
General rebuild #3
-
Start from the 07 model
-
Looking at all difference density peaks above 5.0 rmsd:
-
place water B 1 A 1007 close to 619 HOH and 3.2 angs from N 351.
-
place water B 2 A 1008 between O 240 and N 240.
-
place water B 3 A 1009 between OG1 THR 103 and O 218
-
place water B 4 A 1010 3.0 Angs from N 261 (will need checking).
-
place water B 5 A 1011 3.0 Angs from N 50 (will need checking).
-
place water B 6 A 1012 close to OD2 ASP 352 and slightly far away from N 349
-
place water B 7 A 1013 between O 256 and O 159
-
place water B 8 A 1014 between N 161 and HOH 1013 (one before). Slightly close to CE MET 166
-
182 SER had accidentally deleted the OG atom! put it pack.
-
place water B 9 A 1015 between OD1 ASP 162 and NH1 ARG 165 (ED clearer than the ARG)
-
place water B 10 A 1016 between O 340 and O 627
-
Adjacent water HOH 681 is a big blob in 2Fo-Fc and has 4.7 rms difference density makes distant contacts with NH groups and has a LYS near. Likely to be anion, crystallization conditions in Scapin et al 2013 mention NaCl and MgCl2 so try it as a chloride. CL A 2001 shape is a bit tetrahedral but there is no phosphate mentioned
-
-
Look at buster-report MolProbity side chain rotamer outliers:
-
51 VAL alter to m 20 % (weak ED)
-
127 LEU change to mt 59% - difference density for change
-
307 LEU change to mt 59%
-
388 GLU stub to CB side chain weak
-
137 GLU stub to CB no ED for side chain
-
201 CYS leave alone
-
-
Coordinates after build: 1pmq_07_rebuild2-coot-4_edit.pdb
-
re-refine this as 08 .BUSTER re-refinements (up to 08)
run id |
01 |
02 |
07 |
08 |
description |
MapOnly run to analyse 1pmq.pdb (from tutorial) |
BUSTER refinement of 1pmq.pdb (from tutorial) |
chop out side chains and complete residues with no ED |
place 10 waters and a chloride, sort out MolProbity rotamers |
script |
||||
buster-report |
||||
Rwork |
0.2231 |
0.2062 |
0.2027 |
0.1997 |
Rfree |
0.2619 |
0.2362 |
0.2298 |
0.2256 |
100*(Rfree-Rwork) |
3.9% |
3.0% |
2.7% |
2.6% |
MolProbity Score/percentile |
2.23 / 71st |
1.22 /100 |
1.13 / 100 |
1.00 / 100th |
MolProbity Poor Rotamer |
11 / 5.56% |
8 / 2.59% |
6 / 2.13% |
2 / 0.71% |
MolProbity Ramachandran outliers |
3 / 0.88% |
2 / 0.58% |
0 / 0% |
0 / 0% |
MolProbity Ramachandran favoured |
321 / 93.86% |
328 / 95.91% |
312 / 96.59% |
312 / 96.59% |
-
Statistics good 0.4% drop in Rfree. MolProbity issues fixed …
final rebuild and refine (hopefully)
-
Check model and ED for waters and chlorides.
-
All 16 new waters fine
-
"chloride" this has good ED but distant contacts 3.82 to H 343 and 4.15 to N 283, 3.80 to HOH 627.
-
Is this reasonable? Quick conquest search for NH close to Cl- shows distances N to Cl of 3.0 to 3.5 Angstroms. So these distances are long. Conclusion leave as element type Cl but mark as UNX "unknown atom or ion"
-
Molprobity poor rotamers are A 51 VAL and A 201 CYS. No fix possible
-
373 PRO ED has appeared to allow improvement, real space refine and manuall tweak into difference density.
-
Coot check waters. 621 and 622 have distant contacts and difference density Mark up as UNX rather than waters
-
-
Now lets get back and look at the ANP 2nd ligand:
-
Contour at 0.9 rms, ED now looks interpretable and the sugar ring looks as if it could be tweaked into density.
-
load BUSTER distributed ANP.cif into coot from $BDG_home/tnt/data/common-compounds/ANP.cif
-
turn on torsion restraints in coot using R/RC button
-
coot had glitch on direct real space refine for 502 so copy it as a separate fragment.
-
Real space refine and drag into density:
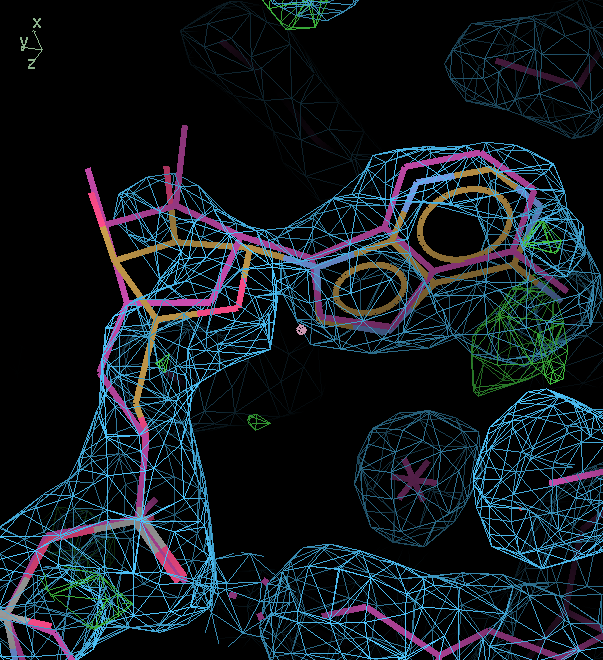
mauve lines: start, orange/atom color after tweak. The sugar ring now better fits ED particularly for atom C5' joined to the phosphate. The adenine ring is also better placed in density. Coordinates for just the ANP after 1pmq_08_rebuild3-coot-1_remodelANP.pdb
-
-
Use an editor to:
-
replace ANP with the new conformation
-
To mark up CL A 2001 as UNK UNX atom to show ambiguity (follow format in http://www.rcsb.org/pdb/files/1AQN.pdb)
-
Move HOH 621 and 622 as to be UNK UNX CL type atoms
-
-
Then "hydrogenate" the ANP because I think ligands best have explicit zero occupancy hydrogen atoms:
hydrogenate -p 1pmq_08_rebuild3-coot-1_edit.pdb -o 1pmq_08_rebuild3-coot-1_edit_hydrogenate_ANP.pdb -l $BDG_home/tnt/data/common-compounds/ANP.cif -ligonly -zero
-
finally remove hydrogen atom HOG2 from the phosphate group because do not its protonation state/which atom.
-
result of all this is 1pmq_08_rebuild3-coot-1_edit_hydrogenate_ANP_edit.pdb
-
start from 1pmq_08_rebuild3-coot-1_edit_hydrogenate_ANP_edit.pdb
-
re-refine from this link:1pmq_09_final1.sh as 09:
run id |
01 |
02 |
08 |
09 |
description |
MapOnly run to analyse 1pmq.pdb (from tutorial) |
BUSTER refinement of 1pmq.pdb (from tutorial) |
place 10 waters and a chloride, sort out MolProbity rotamers |
correct 373, mark two additional "chlorides", tweak ANP conformation |
script |
||||
buster-report |
||||
Rwork |
0.2231 |
0.2062 |
0.1997 |
0.2005 |
Rfree |
0.2619 |
0.2362 |
0.2256 |
0.2249 |
100*(Rfree-Rwork) |
3.9% |
3.0% |
2.6% |
2.4% |
MolProbity Score/percentile |
2.23 / 71st |
1.22 /100 |
1.04 / 100th |
1.04 / 100th |
MolProbity Poor Rotamer |
11 / 5.56% |
8 / 2.59% |
2 / 0.71% |
2 / 0.71% |
MolProbity Ramachandran outliers |
3 / 0.88% |
2 / 0.58% |
0 / 0% |
0 / 0% |
MolProbity Ramachandran favoured |
321 / 93.86% |
328 / 95.91% |
312 / 96.59% |
312 / 96.59% |
check 09 structure/metrics etc.
-
In coot density and placement for ANP looks good at 0.8 rmsd
-
buster-report Mogul results on …-C4'-C5'-… torsion shows that tweak has fixed this from being a "rare" dihedral in 08 structure to "common" so geometry measures back up the change to the ANP.
-
373 PRO tweak is good.
-
check the three UNX UNK CL atoms. All OK’ish and ambiguity is marked well. Idea is to stay there is something here but I am not sure what!
-
Is the 09 structure is ready for re-deposition?
-
As a check look at wwPDB validation reports.
-
For 1pmq.pdb report is Report for 1pmq
(downloaded from
http://www.rcsb.org/pdb/d/file?b=validation&n=1pmq_full_validation.pdf.gz) -
For 09 structure use the wwPDB validation server at
http://wwpdb-validation.wwpdb.org/validservice/
result report for 09 re-refined
-
-
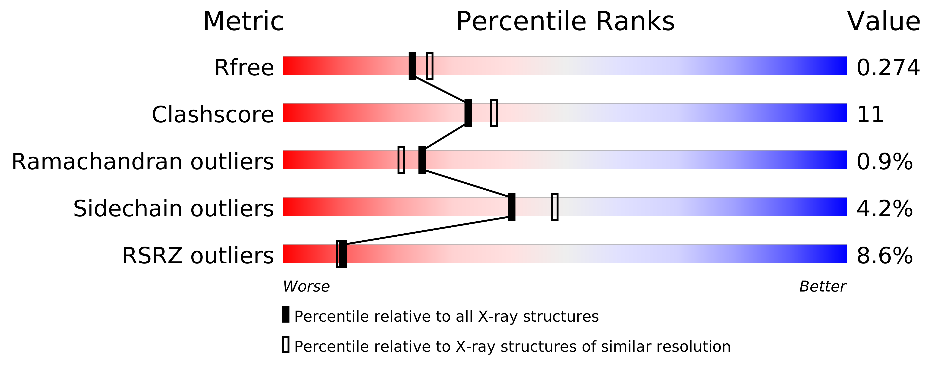
Change in validation sliders:
-
before:
-
after:
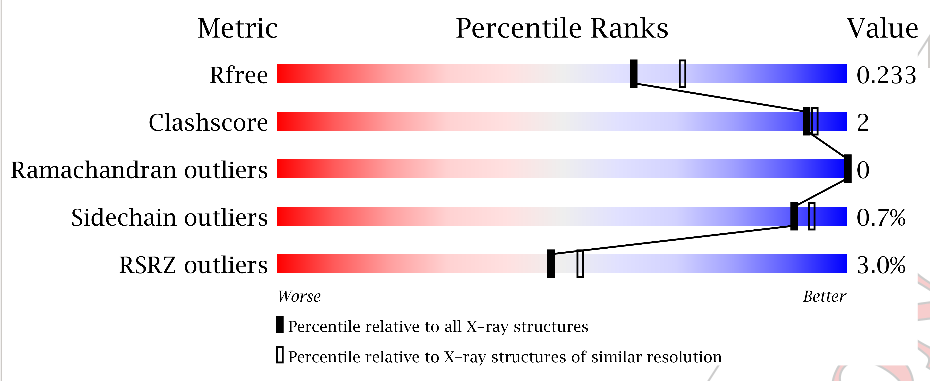
-
-
There are no great issues in the 09 validation report
-
But looking at the buster-report ligand page for the ANP ligand the bond angles and restraints for the phosphoaminophosphonic acid have some issues as assessed by Mogul. The reason is that the ANP dictionary distributed with BUSTER used an old version of grade that did not handle phosphate groups properly. Recent versions of grade do a much better job. So the ANP dictionary that is distributed with BUSTER was updated.
re-refine using an up-to-date ANP dictionary
-
Job 10 is a re-refinement of 09 result using a BUSTER with updated ANP dictionary from this shell script for this 1pmq_10_improvedANP_final2.sh
run id |
01 |
02 |
09 |
10 |
description |
MapOnly run to analyse 1pmq.pdb (from tutorial) |
BUSTER refinement of 1pmq.pdb (from tutorial) |
correct 373, mark two additional "chlorides", tweak ANP conformation |
use up to date ANP |
script |
||||
buster-report |
||||
Rwork |
0.2231 |
0.2062 |
0.2005 |
0.1997 |
Rfree |
0.2619 |
0.2362 |
0.2249 |
0.2258 |
100*(Rfree-Rwork) |
3.9% |
3.0% |
2.4% |
2.6% |
MolProbity Score/percentile |
2.23 / 71st |
1.22 /100 |
1.04 / 100th |
1.07 / 100th |
MolProbity Poor Rotamer |
11 / 5.56% |
8 / 2.59% |
2 / 0.71% |
2 / 0.71% |
MolProbity Ramachandran outliers |
3 / 0.88% |
2 / 0.58% |
0 / 0% |
0 / 0% |
MolProbity Ramachandran favoured |
321 / 93.86% |
328 / 95.91% |
312 / 96.59% |
311 / 96.28% |
-
The alteration in refinement MolProbity metrics are the noise that can be expected.
-
For the ANP ligand the re-refinement has the desired effect sorting out the Mogul outliers noted in 09: see 10 buster-report ligand page for the ANP ligand
-
Use the wwPDB validation server at
http://wwpdb-validation.wwpdb.org/validservice/
result Full wwPDB X-ray Structure Validation report for 10 re-refined -
Note that the "ligand geometry bond angle outliers" in the report are questionable as they are the result of running Mogul with generalization on.
-
-
Change in validation sliders:
-
1pmq.pdb before:
-
model 10:

-
Redeposition
-
the 10 model is now ready for re-deposition to the PDB to obsolete 1pmq.
-
An important thing is record the reason for the replacement this can best be done in the REMARK 900 file that gives information on RELATED ENTRIES. Draft suitable text for this here:
REMARK 900 RELATED ENTRIES REMARK 900 RELATED ID: 1PMQ RELATED DB: PDB REMARK 900 This entry represents a re-refinement of PDB entry 1PMQ. The REMARK 900 structure factors were treated using the Diffraction Anisotropy REMARK 900 Server. The 880 and ANP ligands have been remodelled to achieve REMARK 900 improvement to both stereochemistry and the fit to electron density
-
The model has been re-deposited to the PDB and will be available as 4Z9L
|
Note
|
Page by Oliver Smart from Nov 2014, last updated April 2015. |