Workshop prepared for the Structural Basis of Pharmacology:
Deeper Understanding of Drug Discovery through Crystallography Meeting
Erice June 2014
1. Introduction
-
This tutorial is intended to show how to use BUSTER, grade and buster-report with a particular focus on the ligand page.
-
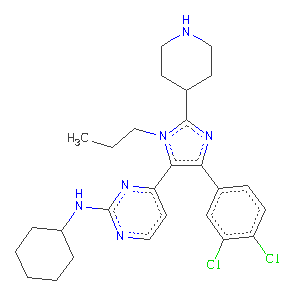
As an example we will use 1pmq the structure of a JNK3 kinase complex, solved in 2003 by Giovanna Scapin at Merck, see 1pmq RCSB. The kinase has been solved in complex with an imidazole-pyrimidine inhibitor:
-
The ligand has been assigned the PDB three letter code 880 for full details visit http://ligand-expo.rcsb.org/reports/8/880/
-
Correcting the ligand placement provides several interesting lessons.
2. Check BUSTER installation
-
First let us check that all required tools are setup properly.
-
This tutorial needs BUSTER refine, grade_PDB_ligand, hydrogenate, buster-report and Mogul to be installed and setup properly.
-
You can check that everything required is OK by running:
checkdeps
-
If there is a problem ask for help!
3. Getting started
-
BUSTER commands are run on the linux or OSX command line.
-
If you are new to linux then visit Learn linux in 10 minutes website
-
The BUSTER reference card lists the most important commands and options
-
Unless you have been given a copy, please print buster_reference_card.pdf now.
-
Then create a fresh directory in which to run the tutorial by using these linux commands
cd mkdir bustertut cd bustertut
4. Starting files
-
A tool fetch_PDB is provided with BUSTER to automatically download the PDB model and structure factors for a given pdb entry. The tool is run from the linux command line by:
fetch_PDB 1pmq
-
The tool uses CCP4 tools to convert the structure factors into mtz format suitable for BUSTER
-
In this case number of warning are produced fetch_PDB 1pmq text output
-
If there are network or firewall problems, then use files from the table below
5. Create grade restraint dictionary for 880 ligand
-
As 1pmq.pdb has the ligand 880 bound it is necessary to create a grade restraint dictionary. If you have a CSDS installation, do this by running on the command line:
grade_PDB_ligand 880
-
This command takes around 3 minutes on a slow single-processor computer. For the impatient download:
-
CIF restraint file |
|
grade "ideal" coordinates |
|
grade output |
-
As the grade output suggests the restraint dictionary for 880 can be examined using the command:
EditREFMAC 880.grade_PDB_ligand.cif 880.grade_PDB_ligand.pdb 880
-
If you do not have CSDS installed you could use the Grade Web Server http://grade.globalphasing.org instead. This produces a results page (this is a local copy).
|
|
For new ligands use a SMILES string input. But existing PDB ligands have defined atom names so use the PDB three-letter residue-type code |
6. Analyze 1pmq.pdb with a BUSTER MapOnly run including -report
-
It is a really good idea to include hydrogen atoms for ligands bound to proteins as this makes the chemistry clear. At this data resolution (2.2Å) I would recommend adding hydrogen atoms to the ligand only and setting their occupancy to zero. This can be quickly done by:
hydrogenate -p 1pmq/1pmq.pdb -o 1pmq_hydrogenate_880.pdb -ligonly -zero -l 880.grade_PDB_ligand.cif
|
|
Q: How do I find the command needed? A: Look at the BUSTER reference card Q: How do I quickly find out about the options for the command? A: Run the command with -h: hydrogenate -h |
-
Then run a BUSTER refine job to calculate Maps (the -M MapOnly option) with buster-report (the -report option).
refine -p 1pmq_hydrogenate_880.pdb -m 1pmq/1pmq.mtz -d 1pmq_01_MapOnly \ -l 880.grade_PDB_ligand.cif -M MapOnly -report >& 1pmq_01_MapOnly.log
-
This command takes around 2 minutes on a slow single processor computer. For the impatient download: 1pmq_01_MapOnly.report.tar.gz (once downloaded unpack it with tar xf 1pmq_01_MapOnly.report.tar.gz)
-
-
Once the MapOnly job has run, look at the buster-report output by:
firefox 1pmq_01_MapOnly.report/index.html
-
Or if pressed for time follow this 1pmq_01_MapOnly.report/index.html link.
-
Does the buster-report output give there any indications of geometry problems?
-
Follow the Ligand analysis page. The electron density for the 880 is clear and the ligand fits it very well:

-
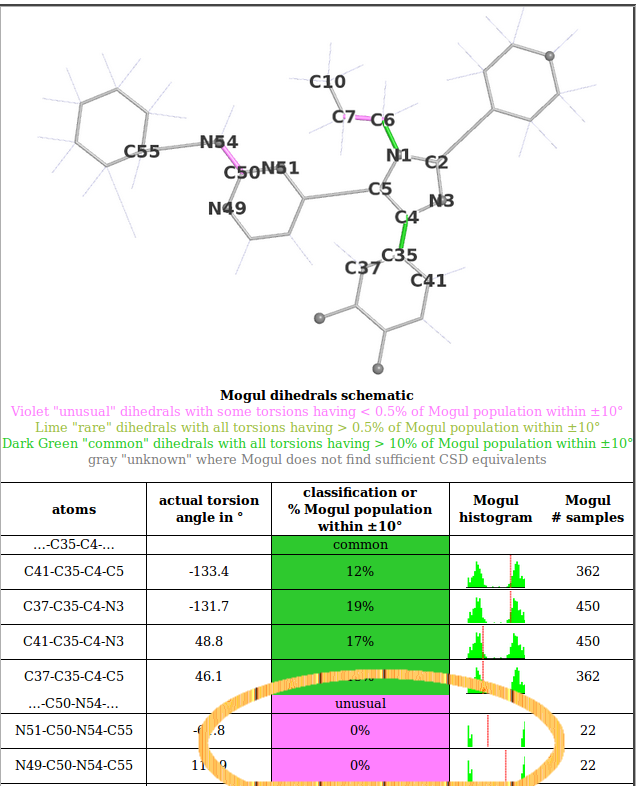
However, the Mogul geometry analysis results show that the fit to electron density is at the cost of distorting the structure compared to what is found in CSD small molecule structures.
-
Detailed ligand analysis page for 880. In particular look at the "Mogul dihedral results". The dihedral …-C50-N54-… is marked as /"unusual"/. The Mogul results strongly suggest that torsion angles N51-C50-N54-C55 and N49-C50-N54-C55 want to be 180 or -180 degrees. This means that atom C55 should be coplanar to the pyrimidine ring but in the 1pqm.pdb structure it is completely out of plane:
-
A really great way to use Coot to look at the structure and map from the report directory is to run our tool:
visualise-geometry-coot 1pmq_01_MapOnly.report/
-
Use the "Geometry issues" tab to find the "Most unhappy" atoms, and note that the problematic section of 880 shows up.
-
-
7. Does BUSTER refinement fix the ligand problem?
-
Run a BUSTER refinement from the PDB structure:
refine -p 1pmq_hydrogenate_880.pdb -m 1pmq/1pmq.mtz -d 1pmq_02_refine \ -l 880.grade_PDB_ligand.cif -M TLSbasic >& \ 1pmq_02_refine.log
-
Oppps! We forgot to run buster-report as part of refine command but this is not a problem as it can be run separately:
buster-report -d 1pmq_02_refine
-
The two commands takes around 30 minutes on a slow single processor computer. So it may be best to download: 1pmq_02_refine-report.tar.gz (once downloaded unpack it with tar xf 1pmq_02_refine-report.tar.gz)
-
Or if pressed for time follow this 1pmq_02_refine-report/index.html link.
-
-
Look at the report using
firefox 1pmq_02_refine-report/index.html
-
Look at examine the RecipSCC plot, and the help page provided. Q: What can you tell about the data and what can be done about it?
|
|
The diffraction is anisotropic and the UCLA MBI — Diffraction Anisotropy Server could help. |
-
Compare results to those for the 01 MapOnly run. In particular Notice the improvement in RecipSCC graph and MolProbity scores.
-
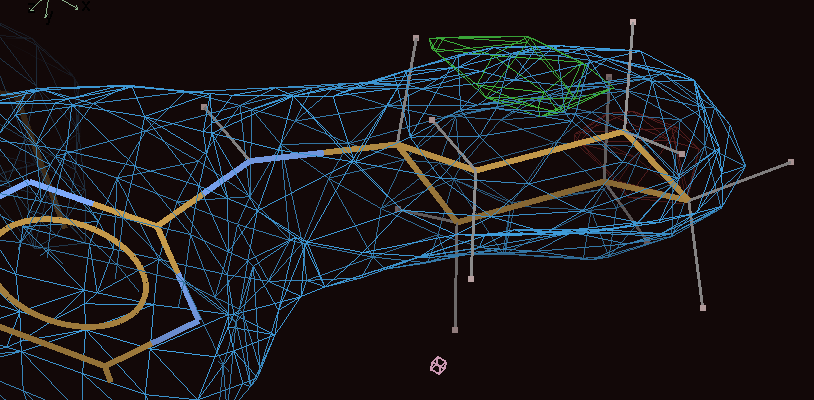
The ligand geometry has improved but both buster-report Mogul angle analysis (1pmq_02_refine-report/ligand/detailedreport_A_501_.html#atableANGLE) and visualise-geometry-coot show there are problems around C54.
-
The problem is that the cyclohexyl ring is the wrong way around and simple refinement cannot fix this.
8. Manual fix of the ligand problem and short re-refinement
-
Start Coot by using our tool to load coordinates, maps and dictionaries:
visualise-geometry-coot 1pmq_02_refine-report/
-
The problem can be sorted by using Coot to rotate the cyclohexyl ring around the 180 degrees around N54-C55 and then applying real-space refinement:
|
|
Use the Torsion General button and click on atoms C50, N54, C55 and either C56 or C60. Before using Real space refine zone click on the R/RC button and tick the Use torsion restraints option. This means that Coot will use grade torsion restraints that are meant to be taken seriously. |
problem |
|
after manual tweak in Coot |
|

-
My coordinates after the tweak: 1pmq_02_refine.report-coot-0.pdb
-
Then do a short re-refine including a buster-report analysis
refine -p 1pmq_02_refine.report-coot-0.pdb -m 1pmq/1pmq.mtz \ -d 1pmq_03_short -l 880.grade_PDB_ligand.cif \ -M ShortRunVoid -report >& 1pmq_03_short.log
-
This command takes around 3.5 minutes on a slow single processor computer. For the impatient download: 1pmq_03_short.report.tar.gz (once downloaded unpack it with tar xf 1pmq_03_short.report.tar.gz)
-
Or if pressed for time follow this 1pmq_03_short.report/index.html link.
-
|
|
there is no need for a full refinement as only a small change had been made to a structure that had already been refined by BUSTER |
-
the report and visualise-geometry-coot show that the cyclohexyl ring is now well positioned in the electron density and has good geometry as assessed by Mogul. The geometry of the hydrogen-bond interaction made by the adjacent NH to a main chain carbonyl has also been improved.
-
But there is some difference density near the chlorine atoms:
-
this looks very much like the ring is adopting two alternates.
-
9. Further work
-
Note that improving the crystallographic model is important to bring out further interpretable detail. The better the model the better the maps it produces. So modeling detail away from the ligand is important. Rfree is a useful validation criterion to make sure that one is not interpreting noise.
-
There are a number of things to look look at the structure:
9.1. Build alternates for the dichlorophenyl ring

-
The density around the ligand from the 03 run shows that there is difference density near the dichlorophenyl ring. The patch of negative density on top of atom CL45 suggests that this position is not fully occupied. On the other side of the ring there is a patch of positive difference density next to H40. This suggests that ring has two alternates. How to deal with this is covered in another tutorial on the BUSTER wiki http://www.globalphasing.com/buster/wiki//index.cgi?AutoBusterLigandAndOccRef
9.2. Examine difference density features in Coot
-
Use Coot options Validate, Difference Map Peaks with Measures, Pointer distances and set the maximum distance to 3.1 Angstroms. Most peaks are water so add them. Think about the others.
9.3. Should 111 GLU be modeled with two alternates?
-
Water HOH 639 looks suspicious. The electron density looks like the GLU could be in two ways around.
9.4. Look at the ANP ligand
-
There is an ANP modeled at 0.5 occupancy near the ligand. Use omit maps to examine the interpretation.
10. Further work: model answer the redeposition of 1pmq
How well did you do in improving the 1pmq model? Producing a "final" structural model for a protein ligand complex is laborious and very often one is interesting features are just at the edge of interpretability. You may be interested in the steps taken to prepare a revised 1pmq model for redeposition to the PDB: notes taken in rebuilding.
|
|
Page by Oliver Smart original version June 2013, last updated April 2015. Address problems, corrections and clarifications to buster-develop@globalphasing.com |